

A Faculdade Inspirar anuncia a parceria com o Instituto Brasileiro de Osteopatia (IBO). Essa colaboração representa um compromisso com a qualidade educacional e com o desenvolvimento profissional de nossos alunos, preparando-os para se destacarem na área. Confira as vantagens para nossos estudantes de Osteopatia
O FUTURO DA FISIOTERPIA PÉLVICA ESTÁ AQUI!
Faça parte do congresso que irá
revolucionar sua abordagem em
Fisioterapia Pélvica.
Modalidades de Cursos




Conheça a Inspirar

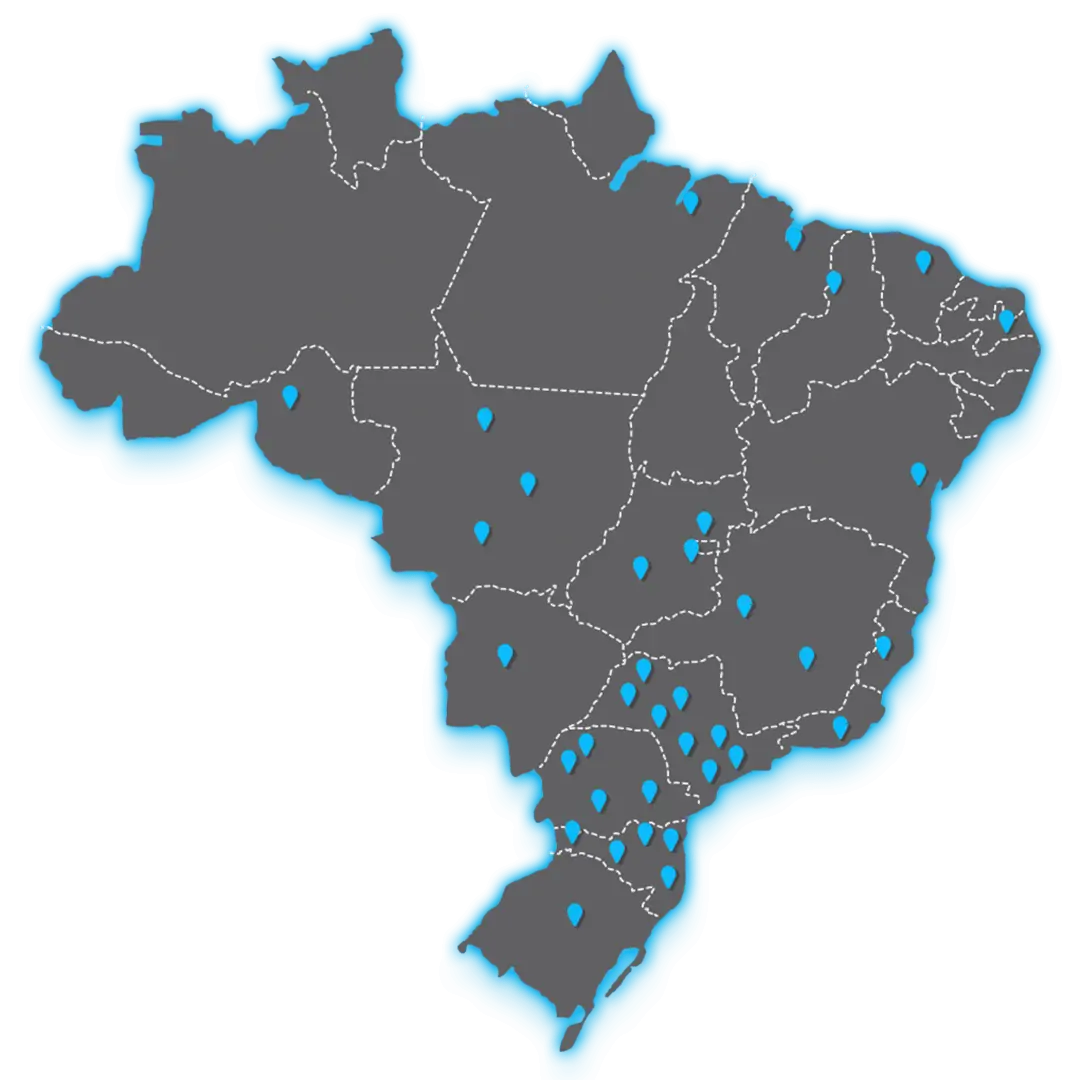
Há mais de duas décadas no mercado, o Grupo Inspirar está presente em todas as regiões do país, além de uma sede em Portugal e outra na Holanda, transmitindo com dedicação e excelência o conhecimento a seus alunos. A Inspirar destaca-se como Instituição referência de ensino na área da Saúde, com cursos de Graduação, Pós-graduação, Extensão e programas de Mestrado e Doutorado. Além disso, é reconhecida como a maior e melhor rede de franquias de pós-graduação do Brasil, segundo a Serasa Experian e Pequenas Empresas & Grandes Negócios.

Mais de 40 unidades
A Inspirar é a maior rede de franquias de cursos voltados à área da Saúde no Brasil.

Mais de 1.000 cursos
Encontre o curso ideal para suas necessidades.

Referência no ensino da área da Saúde
A Faculdade Inspirar já formou milhares de profissionais no mercado de trabalho.
Mais de 25 anos na educação
Já são mais de duas décadas dedicadas à educação do mais alto nível.
Ensino com Reconhecimento
Eleita por 8 anos consecutivos pela Revista Pequenas Empresas & Grandes Negócios uma das 5 principais franquias em cursos e treinamentos do Brasil.

Tradição na Área da Saúde
Com sua origem na área da Saúde, a Inspirar possui a expertise e o know-how necessário para formar um profissional diferenciado para o mercado.

Atendimento Diferenciado
Inspirar e encantar nossos alunos faz parte do dia a dia aqui na Faculdade Inspirar.

Prática e Inovação
A Inspirar busca excelência ao fornecer uma base sólida de conhecimento teórico e prático, incentivando a capacidade de inovação e adaptação.

Conheça a unidade mais próxima a você e venha para a Inspirar.





